WEGO (Web Gene Ontology Annotation Plot) is a simple but useful tool for visualizing, comparing and plotting GO (Gene Ontology) annotation results. As the GO vocabulary was more and more popular, WEGO became widely adopted and used in many researches. Therefore, we have updated WEGO 2.0 in 2018.
The changes made in this update are as following:
1. The limit of input file numbers was cancelled. Now the users could upload as many files as they want in one operation.
2. We have added the reference data of 9 species for the users to choose.
3. Besides the traditional WEGO histogram, WEGO 2.0 outputs an additional type of bar graph, showing GO terms with significant gene number differences.
Quick Start
[Input of WEGO]
WEGO 2.0 supports 5 kinds of input format: WEGO native format, InterProScan text, raw and XML output formats and Gene Ontology Annotation format(GAF). WEGO native format (Fig1) is a simple text file with one annotation record per line. Each column is tab-delimited. The first column is the gene name and the others is the GO. The gene name can be a standard gene identifier, accession number or user’s defined string. The annotation columns can be empty if there is no annotation result available for the gene. One gene can have annotation result in different lines. WEGO will merge the annotation result by gene name. It supports comment line which starts with an exclamation point (!). The samples of WEGO Native file can be downloaded here.
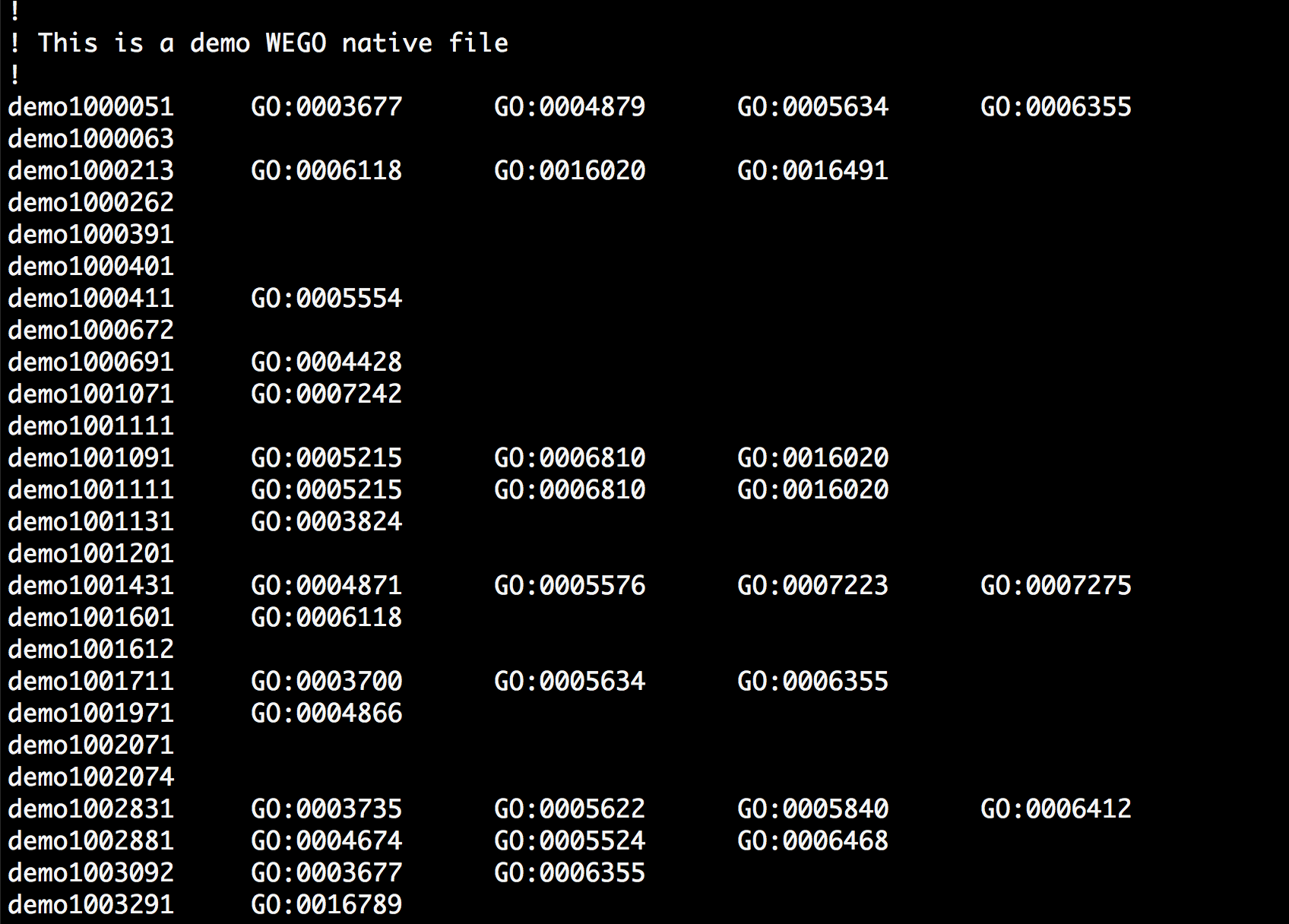
Fig 1. WEGO Native file.
The InterProScan text, raw and XML output formats(https://github.com/ebi-pf-team/interproscan/wiki/OutputFormats) are all acceptable for the convenient using of the users, so that the annotation results of InterProScan can be uploaded onto the WEGO 2.0 without any conversion.
The GO Annotation file format(http://www.geneontology.org/page/go-annotation-file-format-20) is a GAF file which is the default format to upload the annotation result to GO Consortium website. The definition of GO Annotation file format and samples can be found on Gene Ontology website(http://www.geneontology.org/page/download-annotations).
[Uses of WEGO]
There are two ways to work with WEGO 2.0. The first is to upload the annotation file(s). The input file(s) must be in one of the 5 formats described above. The version of Gene Ontology file is optimal for it is suggested to be the same version of what is used in annotation. Also you can choose a reference dataset from the reference list.
The second way is to enter the job ID which was presented during previous analysis on WEGO 2.0 website. The job ID will be available within three days. WEGO 2.0 allows users to change almost all of the settings from their prior session, via this job ID. Even the version of Gene Ontology files can be changed without re-uploading the input files.
[GO Tree View]
GO Tree is shown with a hierarchical GO tree, in which all the GO terms are contained in the uploaded files. Each line(Fig2) of the GO tree represents a GO term. From left to right of each line, is a selection accelerating toolbar, gene number associated to this GO term, gene percentage of the GO term to the uploaded dataset, Pearson Chi-Square test p-value of every input data, GO ID and GO term description, and gene list link to this GO term. Gene percentage is the percentage of the number of the gene with annotation of this term or the term’s child node to the total gene number of a uploaded dataset. If there is only one input data, the Pearson Chi-Square test p-value will not be shown. If any of the expected gene counts are less than 5, the p-value will be shown as ‘Ml’, which stands for meaningless. Star symbol is used to mark the GO term with significant relationship (p-value<0.05) in the input files at the end of the line.
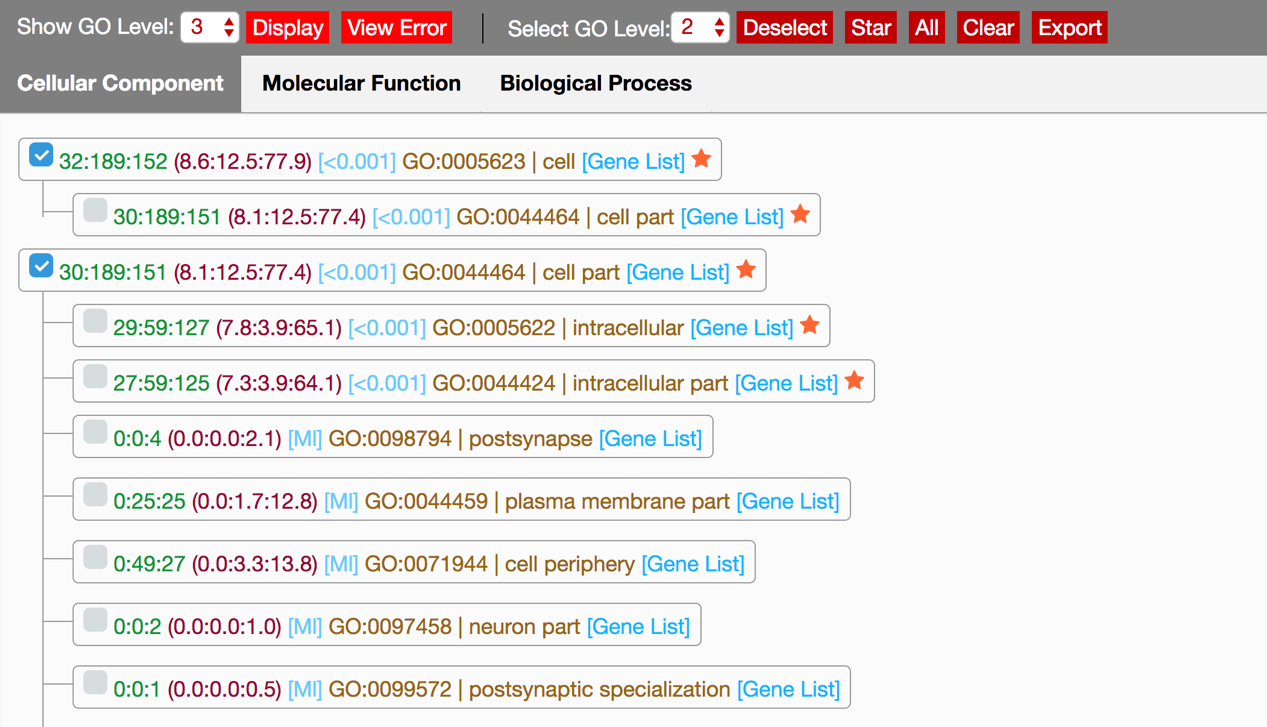
Fig 2. GO tree view after uploading GO annotation file(s).
[View Error]
View Error button helps user to find the GO term in the uploaded file(s) which is not presented in the select Gene Ontology files. It is important to write down the Gene Ontology files version during the gene annotation analysis, so that all GO terms will be included in the downstream statistics. The user can use GO Archive Query tools to help find the right Gene Ontology files when using WEGO 2.0.
[Output of WEGO]
SVG is the default output format of WEGO 2.0, for its wide support by many industrial and open source software, such as Corel DRAW, Illustrator, Inkscape, ImageMagick and so on. With the help of a SVG plug-in, SVG graph could be viewed in a browser. Another advantage of SVG is easy conversion to other graph formats and suitability for publishing. WEGO 2.0 supports other graph formats, including the bitmap formats PNG and JPEG.
The two types of WEGO outputs shown (Fig 3 & Fig 4) are commonly used in a lot of De novo genome projects [1-4] as well as comparative genome projects [5-6] and De novo transcriptome analysis [7-10].
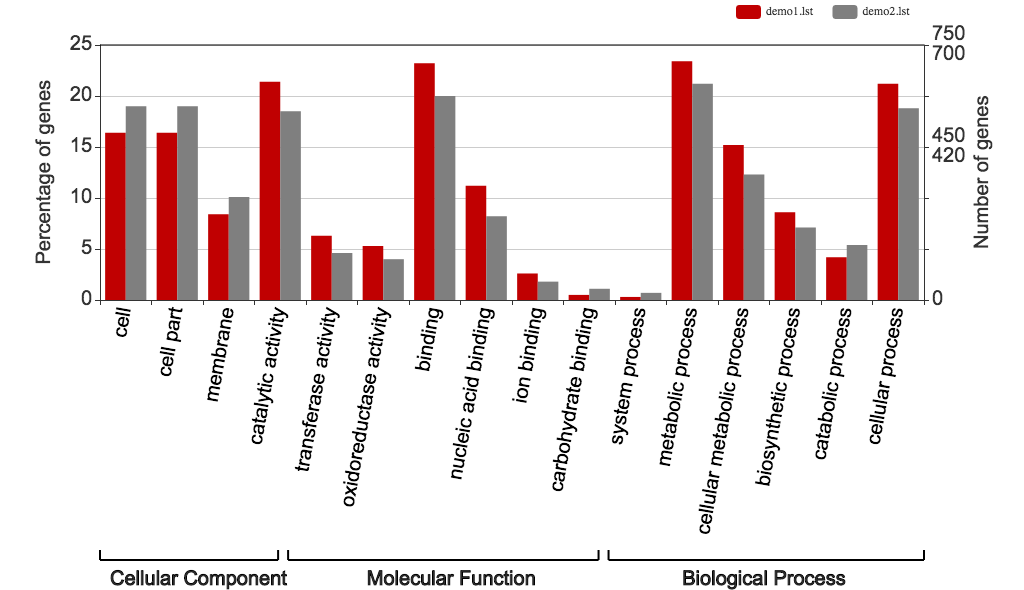
Fig 3. Traditional WEGO histogram. X-axis shows user selected GO terms; y-axis shows the percentages of genes (number of a particular gene divided by total gene number).
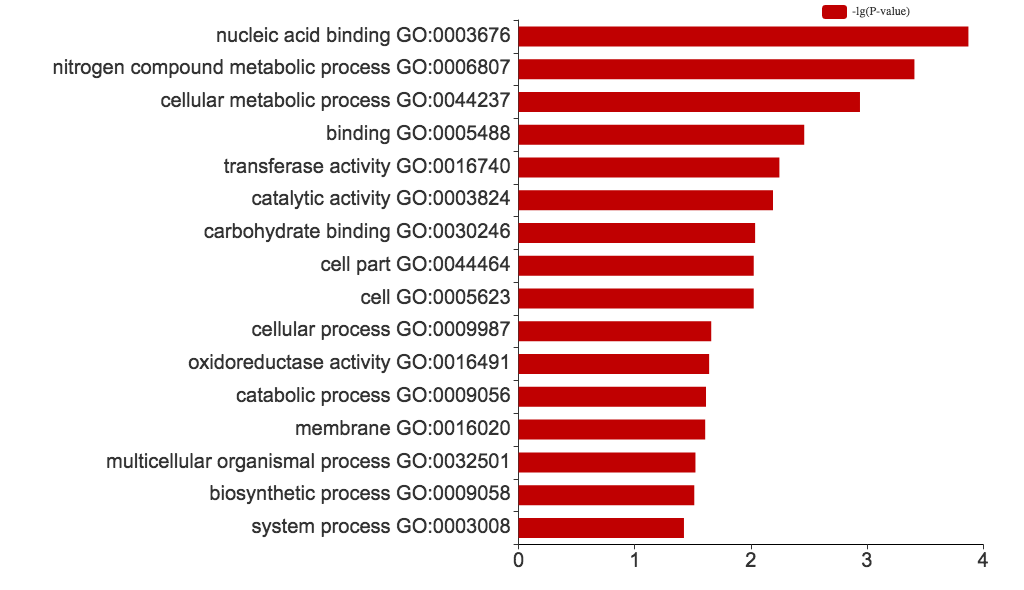
Fig 4. X-axis shows user selected GO terms; y-axis shows the log of the P-values from Chi-square tests (of all the datasets uploaded for a particular GO term).
[External to GO Query]
External to GO Query attempts to make translations between GO and other catalogues of annotation vocabularies. It is an interface based on the database of GO consortium's external2go. We caution that these mapping are neither complete nor exact. The External to GO Query is designed to help biologists to better understand their annotation results. It only deals with the association between GO and the others. Users could query both GO ID and categories of external systems included in the database in External to GO Query. Corresponding entries or GO ID would be given as output. The GO ID could be input in the format of GO:0000015, 0000015 or just 15. The user could choose a special database to search for. Or else, the input will be searched in all external database indexes. Please note that it will take more time.
[GO Archive Query]
GO Archive Query will help the users to find the version of GO Archive where a special GO term exist . With it, the user could choose the proper version of GO Archive to be used in the plotting. The Ontology used in the downstream analysis should be the same as the one used in the annotation. There is a frequently happening error that due to the different version adopted in WEGO analysis and annotation, some GO terms could not be found in the ontology. WEGO will list all of these GO terms in the "View error". And we strongly suggest the user query these GO terms in GO Archive Query, if without the information of the ontology used in the annotation. The GO ID could be input in the format as GO:0000015, 0000015 of just 15. The versions of Gene Ontology containing the GO ID will be given as an output.
Reference:
1. Xia, Q., Zhou, Z., Lu, C., Cheng, D., Dai, F., & Li, B., et al. (2005). Xia q, zhou z, lu c, et al. a draft sequence for the genome of the domesticated silkworm (bombyx mori). science. Science, 306(5703), 1937-1940.
2. Yu, J. Yang H., et al.(2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296(5565), 1937-1942.
3. Xia, Q., Guo, Y., Zhang, Z., Li, D., Xuan, Z., & Li, Z., et al. (2009). Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (bombyx). Science, 326(5951), 433.
4. Wang, L., Tang, N., Gao, X., Chang, Z., Zhang, L., & Zhou, G., et al. (2017). Genome sequence of a rice pest, the white-backed planthopper (sogatella furcifera):. Gigascience, 6(1), 1-9.
5. Li, W., Zhang, L., Ding, Z., Wang, G., Zhang, Y., & Gong, H., et al. (2017). De novo, sequencing and comparative transcriptome analysis of the male and hermaphroditic flowers provide insights into the regulation of flower formation in andromonoecious taihangia rupestris. Bmc Plant Biology,17(1), 54.
6. Krosch, M. N., Bryant, L. M., & Vink, S. (2017). Differential gene expression of australiancricotopus draysoni(diptera: chironomidae) populations reveals seasonal association in detoxification gene regulation:. Scientific Reports, 7(1).
7. Pearce, S. L., Clarke, D. F., East, P. D., Elfekih, S., Gordon, K. H. J., & Jermiin, L. S., et al. (2017). Erratum to: genomic innovations, transcriptional plasticity and gene loss underlying the evolution and divergence of two highly polyphagous and invasive helicoverpa pest species. Bmc Biology, 15(1), 63.
8. Sheng, J., Zheng, X., Wang, J., Zeng, X., Zhou, F., & Jin, S., et al. (2017). Transcriptomics and proteomics reveal genetic and biological basis of superior biomass crop miscanthus. Scientific Reports, 7(1).
9. Wang, Z., Fang, B., Chen, J., Zhang, X., Luo, Z., & Huang, L., et al. (2010). De novo, assembly and characterization of root transcriptome using illumina paired-end sequencing and development of cssr markers in sweetpotato ( ipomoea batatas ). Bmc Genomics, 11(1), 726.
10. Jin, J., Sun, J. B., Park, J. S., Park, Y. K., Arasu, M. V., & Aldhabi, N. A., et al. (2017). De novo transcriptome analysis and glucosinolate profiling in watercress (nasturtium officinale r. br.). Bmc Genomics, 18(1), 401.